Bonjour,
Je vous soumets un problème pratique qui n'a pas reçu de bonne solution. Il s'agit de comparer des "haplotypes". Je vais expliquer brièvement. Pour ceux qui sont pressés l'URL ci-dessous affiche un tableau de valeurs et il faut en déduire une "distance" permettant de situer chaque ligne du tableau, prise globalement, par rapport au groupe.
Qu'est-ce qu'un haplotype ? C'est une mesure génétique faite sur un lot de "marqueurs génétiques". Chacun de ces marqueurs peut prendre toute une série de valeurs (on dit par exemple qu'il y a 8 allèles si le marqueur prend une valeur parmi 8). Le nombre de valeurs (nombre d'allèles) dépend du marqueur. Ces valeurs sont suceptibles de changer à chaque génération et la probabilité d'un changement dépend du marqueur et se mesure par la variabilité (pour un marqueur donné) dans le groupe. Un changement sera considéré comme toujours de 1 (en plus ou en moins). Donc pour prendre un exemple DYS481 étant à 22 il pourra passer (muter) à DYS481=21 ou 23 , bien sûr en général rien ne se passe (pas de mutation) et la valeur reste à 22 (je garde l'exemple de DYS481).
La difficulté qui fait qu'il n'y a pas de bon modèle est qu'il y a des "retour en arrière" possibles et que ceux-ci vont apparaître comme des non mutations alors qu'au contraire il y a eu 2 (ou plus) mutations. Toujours avec l'exemple précédent DYS481, une fois passé à DYS481=23 il est possible que lors d'une génération ultérieure il y ait perte d'une copie (je n'ai pas expliqué : chaque marqueur a des copie répétées et c'est ce nombre de copies qui peut baisser ou augmenter de 1 à chaque fois) ramenant DYS481 à 22 la valeur initiale.
Donc, à partir d'un lot d'individus (les lignes du tableau) dont on connaît les valeurs pour 67 marqueurs (quelques uns ont des valeurs supplémentaires ; ne pas en tenir compte) , chaque marqueur étant en colonne, on veut trouver "l'âge" du groupe (en nombre de générations). Pour cela on suppose qu'un haplotype ayant pour valeur pour chaque marqueur la moyenne observée dans le groupe représente "l'ancêtre" dont sont dérivés les haplotypes observés (ce point peut être contesté mais il est généralement admis). Le groupe que j'ai donné comme exemple est très homogène mais on cherche une méthode permettant d'analyser aussi bien des groupes moins homogènes. Si vous avez des questions n' hésitez pas.
Modéliser les distances entre haplotypes
14 messages
- Page 1 sur 1

![]() par nuage » 22 Mar 2008, 21:04
par nuage » 22 Mar 2008, 21:04
Salut,
je ne comprend pas vraiment ta question. En particulier je n'ai pas vu la ligne DYS481.
Mais je crois comprendre que chaque colonne suit (à une constante près) une loi binomiale de paramètres n (nombre de générations) et p (probabilité de mutation).
Si c'est le cas il est sans doute judicieux d'examiner la colonne où la variance est maximale.
On peut sans doute aussi examiner avec profit la loi de) .
.
Surtout si on a une majoration de p.
Mais encore un fois, je ne crois pas avoir vraiment compris ton problème.
Essaye de donner un exemple figurant dans le tableau.
Et que signifient les en-têtes des colonnes ?
Amicalement,
nuage :
je ne comprend pas vraiment ta question. En particulier je n'ai pas vu la ligne DYS481.
Mais je crois comprendre que chaque colonne suit (à une constante près) une loi binomiale de paramètres n (nombre de générations) et p (probabilité de mutation).
Si c'est le cas il est sans doute judicieux d'examiner la colonne où la variance est maximale.
On peut sans doute aussi examiner avec profit la loi de
Surtout si on a une majoration de p.
Mais encore un fois, je ne crois pas avoir vraiment compris ton problème.
Essaye de donner un exemple figurant dans le tableau.
Et que signifient les en-têtes des colonnes ?
Amicalement,
nuage :
![]() par perplexe » 22 Mar 2008, 22:00
par perplexe » 22 Mar 2008, 22:00
Je fais une réponse rapide pour commencer :
DYS481 est un marqueur et il est en colonne. Chaque colonne (marqueur) est indépendant(e) ; il est possible de travailler sur des haplotypes avec plus ou moins de marqueurs (ici j'ai sélectionné des haplotypes ayant au moins 67 marqueurs en commun). Chaque ligne est un haplotype . Pour la loi de proba modélisant l'accumulation des mutations je pense qu'une loi binomiale est OK.
DYS481 est un marqueur et il est en colonne. Chaque colonne (marqueur) est indépendant(e) ; il est possible de travailler sur des haplotypes avec plus ou moins de marqueurs (ici j'ai sélectionné des haplotypes ayant au moins 67 marqueurs en commun). Chaque ligne est un haplotype . Pour la loi de proba modélisant l'accumulation des mutations je pense qu'une loi binomiale est OK.
![]() par perplexe » 22 Mar 2008, 23:04
par perplexe » 22 Mar 2008, 23:04
Je reprends mes explications. A propos de DYS481 : c'est le 9ème marqueur en partant de la droite (écrit 481 pour simplifier) , la valeur la plus fréquente est DYS481=22 mais les valeurs s'étalent de 19 à 24 (donc pas symétrique). Vous pouvez vous étonner que de nombreux marqueurs varient très peu voire pas du tout (par exemple DYS393 , le premier en partant de la gauche). Ceci a été voulu, précisément pour éviter les cas où plusieurs mutations ramènent à l'état initial. Dans l'exemple fourni par le lien on pourrait penser que ce sont les colonnes où il y a le plus de variation qui sont les plus informatives (576, 481, CDYa, etc..) mais ce sont aussi les cas où le risque de "retour arrière" est le plus élevé. C'est pour cette raison que le choix a été fait de travailler avec un grand nombre de marqueurs, chacun variant peu.
Au delà de mon exemple vous pouvez vouloir inspecter un cas comportant un plus grand nombre d'haplotypes et plus hétérogène. vous trouverez ceci à :
http://www.yseach.org , sélectionnez ensuite "search by haplogroup" ; une liste déroulante vous permet de choisir, par exemple "R1b1c" . Une difficulté est que ce site tend à bloquer sur les URL trop longues : après avoir fait "Check all" (coche tous les haplotypes) puis "compare" il peut arriver que vous ayez un refus d'obtempérer mais peut-être saurez vous trouver la parade. Sinon je possède des fichiers Excel correspondants...
Au delà de mon exemple vous pouvez vouloir inspecter un cas comportant un plus grand nombre d'haplotypes et plus hétérogène. vous trouverez ceci à :
http://www.yseach.org , sélectionnez ensuite "search by haplogroup" ; une liste déroulante vous permet de choisir, par exemple "R1b1c" . Une difficulté est que ce site tend à bloquer sur les URL trop longues : après avoir fait "Check all" (coche tous les haplotypes) puis "compare" il peut arriver que vous ayez un refus d'obtempérer mais peut-être saurez vous trouver la parade. Sinon je possède des fichiers Excel correspondants...
![]() par perplexe » 31 Mar 2008, 17:37
par perplexe » 31 Mar 2008, 17:37
Je vous propose ma solution pour étudier ces haplotypes et j'attends vos critiques. J'utilise 2 fichiers Excel que j'ai chargés sur :
http://www.megaupload.com/?d=6KH5OCUA pour le fichier de départ
http://www.megaupload.com/?d=7LYOEPXY pour le fichier avec traitement stat
Je ne reviens pas sur le principe ; voire les messages précedents. Le fichier contient 2455 haplotypes sur 31 marqueurs ; donc 2455 lignes et 31 colonnes de nombres à traiter.
Voilà comment j'ai traité les données :
- pour chaque colonne (chaque marqueur) la moyenne est calculée puis cette moyenne est ramenée à l'entier le plus proche et le nombre obtenu est ce que j'appelle (ici) valeur modale (ou "modale" dans le tableau). Cette valeur est supposée être la valeur du "fondateur" et les haplotypes de la population sont des dérivés à partir de ces valeurs du fondateur par ajout ou perte d'une unité (le processus pouvant se répéter - pertes de plusieurs unité pour un marqueur donné).
- pour chaque colonne l'écart type est calculé.
- dans chaque colonne je soustrais la valeur modale et je divise par l'écart type.
- pour chaque ligne (haplotype) je fais la somme des carrés des valeurs obtenues ci-dessus . Ceci a l'avantage de supprimer la question du signe et de donner un résultat homogène à une distance mais je ne sais pas bien justifier cette étape par rapport au choix de la somme des valeurs absolues.
Qu'en pensez-vous ?
http://www.megaupload.com/?d=6KH5OCUA pour le fichier de départ
http://www.megaupload.com/?d=7LYOEPXY pour le fichier avec traitement stat
Je ne reviens pas sur le principe ; voire les messages précedents. Le fichier contient 2455 haplotypes sur 31 marqueurs ; donc 2455 lignes et 31 colonnes de nombres à traiter.
Voilà comment j'ai traité les données :
- pour chaque colonne (chaque marqueur) la moyenne est calculée puis cette moyenne est ramenée à l'entier le plus proche et le nombre obtenu est ce que j'appelle (ici) valeur modale (ou "modale" dans le tableau). Cette valeur est supposée être la valeur du "fondateur" et les haplotypes de la population sont des dérivés à partir de ces valeurs du fondateur par ajout ou perte d'une unité (le processus pouvant se répéter - pertes de plusieurs unité pour un marqueur donné).
- pour chaque colonne l'écart type est calculé.
- dans chaque colonne je soustrais la valeur modale et je divise par l'écart type.
- pour chaque ligne (haplotype) je fais la somme des carrés des valeurs obtenues ci-dessus . Ceci a l'avantage de supprimer la question du signe et de donner un résultat homogène à une distance mais je ne sais pas bien justifier cette étape par rapport au choix de la somme des valeurs absolues.
Qu'en pensez-vous ?
![]() par JJa » 01 Avr 2008, 07:16
par JJa » 01 Avr 2008, 07:16
Bonjour,
j'ai eu l'occasion de travailler sur un problème de reconnaissance d'espèce qui semble voisin de celui dont vous parlez. Plus précisément, ma contribution a été de faire la liaison entre d'une part, des modèles de distances non-euclidiennes de points dans l'hyperespace des paramètres et d'autre part, des procédés statistiques classiques, de façon à bénéficier de synergies entre ces deux approches.
Actuellement, je n'envisage pas de revenir à ce domaine d'études. De plus, il est hors de question de communiquer des rapports et logiciels qui ont été réalisés dans le cadre d'une colaboration avec un laboratoire de recherches bien connu.
Néanmoins, si vous prenez contact avec moi par message privé, je pourrais éventuellement répondre à des questions d'ordre général et au besoin, vous comuniquer des références dans ce domaine.
j'ai eu l'occasion de travailler sur un problème de reconnaissance d'espèce qui semble voisin de celui dont vous parlez. Plus précisément, ma contribution a été de faire la liaison entre d'une part, des modèles de distances non-euclidiennes de points dans l'hyperespace des paramètres et d'autre part, des procédés statistiques classiques, de façon à bénéficier de synergies entre ces deux approches.
Actuellement, je n'envisage pas de revenir à ce domaine d'études. De plus, il est hors de question de communiquer des rapports et logiciels qui ont été réalisés dans le cadre d'une colaboration avec un laboratoire de recherches bien connu.
Néanmoins, si vous prenez contact avec moi par message privé, je pourrais éventuellement répondre à des questions d'ordre général et au besoin, vous comuniquer des références dans ce domaine.
![]() par niko14 » 05 Avr 2008, 09:19
par niko14 » 05 Avr 2008, 09:19
Bonjour,
Je ne suis pas sur d'avoir tous lu, mais il me semble que la solution au problème existe déjà.
Le problème que tu cites (retour à un état précédent de l'ADN), est un phénomène bien connu des généticiens (on appelle cela la réversion, ou plus généralement homoplasie, ce qui aboutit à de la "saturation" sur le gène). Il existe des dizaines de modèles d'évolution des séquences d'ADN qui permettent de gérer ces problèmes (voir par exemple http://darwin.uvigo.es/software/modeltest.html).
Cela correspond-il à ton problème ?
Niko
Je ne suis pas sur d'avoir tous lu, mais il me semble que la solution au problème existe déjà.
Le problème que tu cites (retour à un état précédent de l'ADN), est un phénomène bien connu des généticiens (on appelle cela la réversion, ou plus généralement homoplasie, ce qui aboutit à de la "saturation" sur le gène). Il existe des dizaines de modèles d'évolution des séquences d'ADN qui permettent de gérer ces problèmes (voir par exemple http://darwin.uvigo.es/software/modeltest.html).
Cela correspond-il à ton problème ?
Niko
![]() par perplexe » 05 Avr 2008, 23:48
par perplexe » 05 Avr 2008, 23:48
La difficulté à propos du "retour" est effectivement ce qu'on appelle homoplasie mais le problème de comparaison d'haplotypes pour déterminer , à partir d'une population d'haplotypes ayant un "ancêtre" commun, l'âge de ce groupe est un problème bien spécifique. Il ne s'agit pas de substitution de nucléotide mais de gain ou d'ajout d'un "motif" ADN et on repère des nombres de "motifs" répétés. Je n'étais pas rentré dans ces détails parce que je ne voulais pas trop compliquer.
Donc, pour résumer, la difficulté n'est pas limitée à l'homoplasie ; j'avais insisté sur ce point parce que dans le développement d'une solution il faut l'envisager dès le début. Dans le début de traitement des données que j'ai placé dans les fichiers téléchargeables je tente d'homogénéiser les données de tous les marqueurs en divisant par l'écart type et ensuite je peux sommer les mesures de tous les marqueurs d'un haplotype ; je n'ai plus ensuite qu'une seule valeur à comparer entre haplotypes , la somme obtenue. Dans ce cas je n'ai rien tenté pour prendre en compte l'homoplasie. Si quelqu'un a mieux à proposer je suis preneur.
Donc, pour résumer, la difficulté n'est pas limitée à l'homoplasie ; j'avais insisté sur ce point parce que dans le développement d'une solution il faut l'envisager dès le début. Dans le début de traitement des données que j'ai placé dans les fichiers téléchargeables je tente d'homogénéiser les données de tous les marqueurs en divisant par l'écart type et ensuite je peux sommer les mesures de tous les marqueurs d'un haplotype ; je n'ai plus ensuite qu'une seule valeur à comparer entre haplotypes , la somme obtenue. Dans ce cas je n'ai rien tenté pour prendre en compte l'homoplasie. Si quelqu'un a mieux à proposer je suis preneur.
![]() par nuage » 07 Avr 2008, 19:51
par nuage » 07 Avr 2008, 19:51
Salut perplexe,
j'ai essayé de faire un modèle sur le premier lien que tu as donné (je n'arrive pas à lire les autres : on me propose des rendez-vous galants mais pas de télécharger tes fichiers).
Mon idée de base a été la suivante :
Les individus (lignes) sont indépendants et chaque valeur est obtenue en ajoutant ou en enlevant 1 à une valeur de départ, ou en la laissant telle quelle. La proba d'ajouter 1 étant égale à la proba d'enlever 1, sinon le choix de la moyenne pour représenter l'ancêtre est incohérent.
Le problème est que le modèle ne marche pas vraiment bien.
Je crois que je ne suis pas assez compétent pour pouvoir t'aider. Enfin je suis en vacances et je vais encore un peu chercher.
A+
Ps : il y a une valeur aberrante dans ton premier tableau : le marqueur CDYa pour l'individu SP6RT
j'ai essayé de faire un modèle sur le premier lien que tu as donné (je n'arrive pas à lire les autres : on me propose des rendez-vous galants mais pas de télécharger tes fichiers).
Mon idée de base a été la suivante :
Les individus (lignes) sont indépendants et chaque valeur est obtenue en ajoutant ou en enlevant 1 à une valeur de départ, ou en la laissant telle quelle. La proba d'ajouter 1 étant égale à la proba d'enlever 1, sinon le choix de la moyenne pour représenter l'ancêtre est incohérent.
Le problème est que le modèle ne marche pas vraiment bien.
Je crois que je ne suis pas assez compétent pour pouvoir t'aider. Enfin je suis en vacances et je vais encore un peu chercher.
A+
Ps : il y a une valeur aberrante dans ton premier tableau : le marqueur CDYa pour l'individu SP6RT
![]() par perplexe » 07 Avr 2008, 22:56
par perplexe » 07 Avr 2008, 22:56
Nuage, merci de ton retour. Ta compréhension du "sujet" est bonne et je pense que tu vas pouvoir travailler avec un fichier plus complet. J'ai testé le lien avec "megaupload" ; ils ont effectivement introduit des images "galantes" mais c'est téléchargeable : ils demandent de taper des lettres / chiffres en haut dans une case et de confirmer , ensuite il faut patienter 40 secondes car j'ai utilisé leur version gratuite et alors, alors enfin on peut avoir le fichier Excel ! Je vais voir si je ne peux pas trouver mieux mais c'est fonctionnel ... Ces deux fichiers sont à préférer si possible car le nombre d'haplotypes est vraiment "statistique" et ces haplotypes sont ceux du groupe principal en Europe de l'ouest (appelé R1b1c).
Bien vu pour la valeur aberrante ; le mieux est de supprimer la ligne ou de travailler sans ce marqueur pour tout le monde. Il y a aussi un haplotype qui n'a pas toutes les valeurs , là aussi le mieux est de le supprimer. Encore une fois , le mieux est de passer au fichier téléchargé.
A propos du fait de se passer d'un marqueur, il faut bien comprendre que le nombre de marqueurs disponibles sur la population étudiée peut changer et plus il y a de marqueurs plus on peut espérer un bonne ségrégation /précision dans l'étude de ces haplotypes. On essaie toujours de se placer dans le cas où tous les haplotypes qu'on étudie ont été testés pour les mêmes marqueurs quitte à supprimer quelques marqueurs pour avoir une population d'étude plus importante.
Dernière minute : j'ai téléchargé le fichier "R1b1c" avec quelques corrections sur http://rapidshare.de/files/39053834/haplotest31-corrig_.xls.html . Il faut cliquer sur "free" puis attendre 60 secondes (décompte visible) et aussi entrer les chiffres / lettres comme précédemment.
Il n'y a , pour l'instant que le fichier de base et pas le fichier avec un début de traitement des données.
Bien vu pour la valeur aberrante ; le mieux est de supprimer la ligne ou de travailler sans ce marqueur pour tout le monde. Il y a aussi un haplotype qui n'a pas toutes les valeurs , là aussi le mieux est de le supprimer. Encore une fois , le mieux est de passer au fichier téléchargé.
A propos du fait de se passer d'un marqueur, il faut bien comprendre que le nombre de marqueurs disponibles sur la population étudiée peut changer et plus il y a de marqueurs plus on peut espérer un bonne ségrégation /précision dans l'étude de ces haplotypes. On essaie toujours de se placer dans le cas où tous les haplotypes qu'on étudie ont été testés pour les mêmes marqueurs quitte à supprimer quelques marqueurs pour avoir une population d'étude plus importante.
Dernière minute : j'ai téléchargé le fichier "R1b1c" avec quelques corrections sur http://rapidshare.de/files/39053834/haplotest31-corrig_.xls.html . Il faut cliquer sur "free" puis attendre 60 secondes (décompte visible) et aussi entrer les chiffres / lettres comme précédemment.
Il n'y a , pour l'instant que le fichier de base et pas le fichier avec un début de traitement des données.
![]() par nuage » 09 Avr 2008, 16:52
par nuage » 09 Avr 2008, 16:52
Salut,
j'ai pas fait grand chose...
Je te donne quand même la loi de la v.a.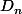 >
>
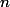 est le nombre de générations, à chaque génération
est le nombre de générations, à chaque génération 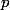 est la probabilité d'un changement
est la probabilité d'un changement 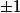 (la proba d'un changement +1 est
(la proba d'un changement +1 est  )
) 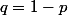 est la proba que rien ne change.
est la proba que rien ne change. )
=P(D_n=-k)=\sum_{i=0}^{a_k}{{k+2 i}\choose{k}}{{n}\choose {k+2 i}} \left(\frac{p}{2}\right)^{2 i}q^{n-2 i}) pour
pour 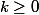
On peut remarquer que :
A part ça j'ai un problème théorique : peut-on légitimement considérer les individus comme indépendants ? Ou faut-il essayer de faire un arbre généalogique ?
Les marqueurs 5 7 6 et 4 3 8 donnent des valeurs étonnantes, je vais donc les supprimer de mes calculs.
A+ sans doute par mp
j'ai pas fait grand chose...
Je te donne quand même la loi de la v.a.
On peut remarquer que :
ce qui permet d'estimer
- pour
du moins pour les valeurs vraisemblables.
A part ça j'ai un problème théorique : peut-on légitimement considérer les individus comme indépendants ? Ou faut-il essayer de faire un arbre généalogique ?
Les marqueurs 5 7 6 et 4 3 8 donnent des valeurs étonnantes, je vais donc les supprimer de mes calculs.
A+ sans doute par mp
![]() par perplexe » 09 Avr 2008, 22:00
par perplexe » 09 Avr 2008, 22:00
Je vais étudier le modèle mais je fais cette réponse rapide pour confirmer que l'indépendance des individus vient de ce qu'il s'agit d'un échantillonnage dans une population beaucoup plus grande (très faible probabilité de tomber sur des parents proches) . Les individus sont tous liés génétiquement mais on ne sait pas comment et ça n'importe pas dans le traitement des données.
14 messages
- Page 1 sur 1
Qui est en ligne
Utilisateurs parcourant ce forum : Aucun utilisateur enregistré et 9 invités
Tu pars déja ?
Fais toi aider gratuitement sur Maths-forum !
Créé un compte en 1 minute et pose ta question dans le forum ;-)
Identification
Pas encore inscrit ?
Ou identifiez-vous :